This vignette documents how dabestr is able to compute
the calculation of delta-delta, an experimental function that allows the
comparison between two bootstrapped effect sizes computed from two
independent categorical variables.
Many experimental designs investigate the effects of two interacting independent variables on a dependent variable. The delta-delta effect size lets us distill the net effect of the two variables. To illustrate this, let’s delve into the following problem…
Consider an experiment where we test the efficacy of a drug named
Drugon a disease-causing mutationMbased on a disease metricY. In this experiment, the greater the valueYis, the more severe the disease phenotype is. The phenotypeYhas been shown to be caused by a gain of the function mutationM, so we expect a difference between the wild type (W) subjects and the mutant subjects (M). We want to know whether this effect is ameliorated by the administration of aDrugtreatment. We also administer a placebo as a control. In theory, we only expect theDrugto have an effect on theMgroup, although in practice, many drugs have non-specific effects on healthy populations as well.
Effectively, we have 4 groups of subjects for comparison:
| Wild type | Mutant | |
|---|---|---|
| Drug | ||
| Placebo |
There are 2 Treatment conditions: the
Placebo (control group) and the Drug (test
group). There are 2 Genotypes: W (wild type
population) and M (mutant population). Additionally, each
experiment was conducted twice (Rep1 and
Rep2). We will perform a few analyses to visualise these
differences in a simulated dataset.
Create dataset for demo
set.seed(12345) # Fix the seed so the results are reproducible.
# pop_size = 10000 # Size of each population.
N <- 20 # The number of samples taken from each population
# Create samples
placebo <- rnorm(N / 2, mean = 4, sd = 0.4)
placebo <- c(placebo, rnorm(N / 2, mean = 2.8, sd = 0.4))
drug <- rnorm(N / 2, mean = 3, sd = 0.4)
drug <- c(drug, rnorm(N / 2, mean = 2.5, sd = 0.4))
# Add a `Genotype` column as the second variable
genotype <- c(rep("M", N / 2), rep("W", N / 2))
# Add an `id` column for paired data plotting.
id <- 1:N
# Add a `Rep` column as the first variable for the 2 replicates of experiments done
Rep <- rep(c("Rep1", "Rep2"), N / 2)
# Combine all columns into a DataFrame.
df <- tibble::tibble(
Placebo = placebo,
Drug = drug,
Genotype = genotype,
ID = id,
Rep = Rep
)
df <- df %>%
tidyr::gather(key = Treatment, value = Measurement, -ID, -Genotype, -Rep)| Genotype | ID | Rep | Treatment | Measurement |
|---|---|---|---|---|
| M | 1 | Rep1 | Placebo | 4.234211 |
| M | 2 | Rep2 | Placebo | 4.283786 |
| M | 3 | Rep1 | Placebo | 3.956279 |
| M | 4 | Rep2 | Placebo | 3.818601 |
| M | 5 | Rep1 | Placebo | 4.242355 |
| M | 6 | Rep2 | Placebo | 3.272818 |
Loading Data
To make a delta-delta plot, you need to simply set
delta2 = TRUE in the load() function. The
colour parameter will be used to determine the colour of
dots for the scattered plots or the colour of lines for the slopegraphs.
The experiment parameter will be used to specify the
grouping of the data. For delta-delta plots, the idx
parameter is optional. Here’s an example:
Unpaired Data
unpaired_delta2 <- load(df,
x = Genotype, y = Measurement,
experiment = Treatment, colour = Genotype,
delta2 = TRUE
)The above function creates the following dabest
object:
print(unpaired_delta2)
#> DABESTR v2025.3.14
#> ==================
#>
#> Good morning!
#> The current time is 07:06 AM on Wednesday July 29, 2026.
#>
#> ffect size(s) with 95% confidence intervals will be computed for:
#> 1. M Placebo minus W Placebo
#> 2. M Drug minus W Drug
#> 3. Drug minus Placebo (only for mean difference)
#>
#> 5000 resamples will be used to generate the effect size bootstraps.We can quickly check out the effect sizes:
unpaired_delta2.mean_diff <- mean_diff(unpaired_delta2)
print(unpaired_delta2.mean_diff)
#> DABESTR v2025.3.14
#> ==================
#>
#> Good morning!
#> The current time is 07:06 AM on Wednesday July 29, 2026.
#>
#> The character(0) mean difference between M Placebo and W Placebo is 1.032 [95%CI 0.731, 1.279].
#> The p-value of the two-sided permutation t-test is 0.0000, calculated for legacy purposes only.
#>
#> The character(0) mean difference between M Drug and W Drug is 0.244 [95%CI -0.136, 0.666].
#> The p-value of the two-sided permutation t-test is 0.2695, calculated for legacy purposes only.
#>
#> 5000 bootstrap samples were taken; the confidence interval is bias-corrected and accelerated.
#> Any p-value reported is the probability of observing the effect size (or greater),
#> assuming the null hypothesis of zero difference is true.
#> For each p-value, 5000 reshuffles of the control and test labels were performed.
dabest_plot(unpaired_delta2.mean_diff)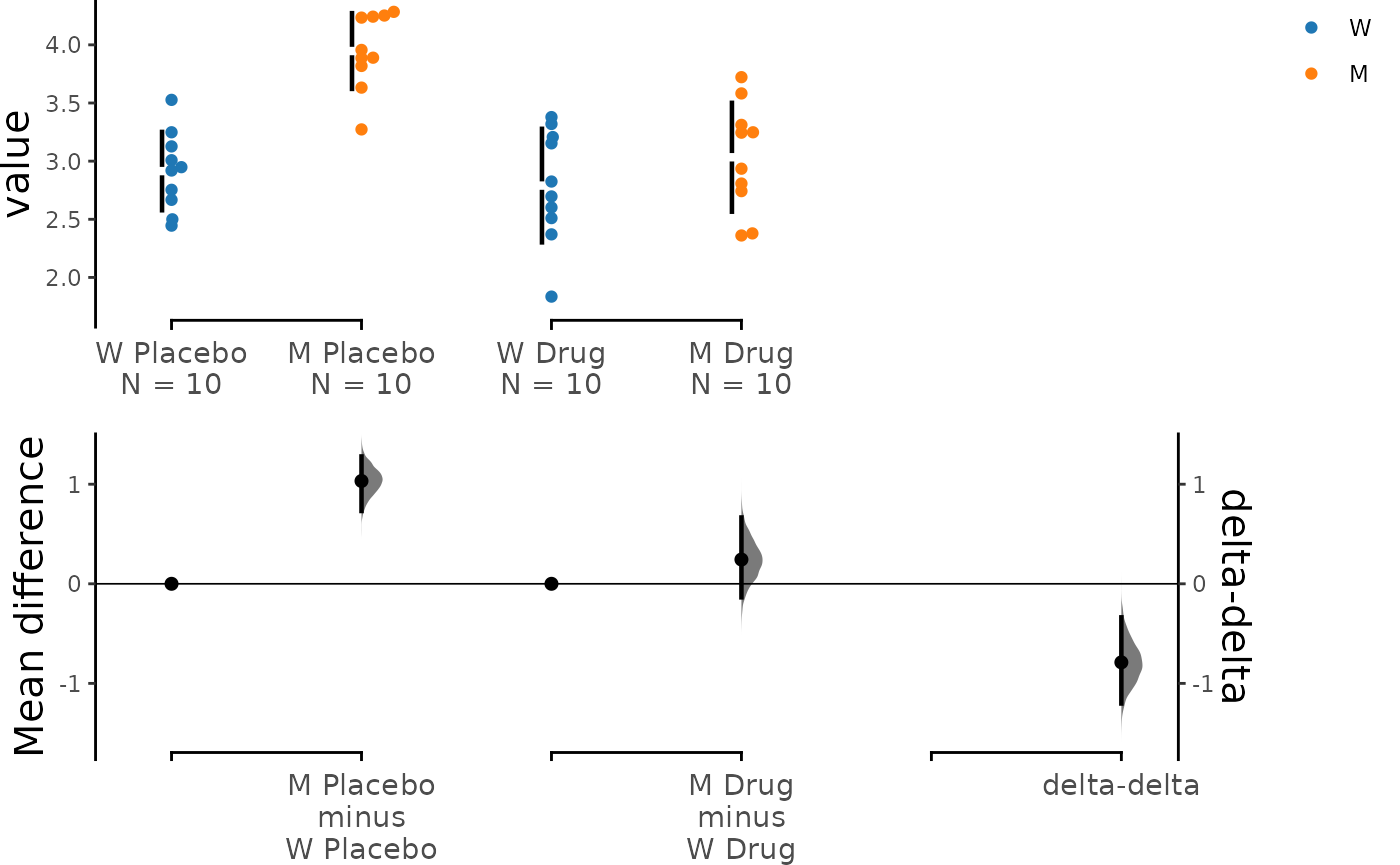
The horizontal axis in the above plot represents the
Genotype condition, and the dot colour is also specified by
Genotype. The left pair of scattered plots corresponds to
the Placebo group, while the right pair is based on the
Drug group. The bottom left axis contains the two primary
deltas: the Placebo delta and the Drug
delta.
It is evident that when only the placebo was administered, the mutant
phenotype was around 1.23 [95% CI: 0.948, 1.52]. However, this
difference was reduced to approximately 0.326 [95% CI: 0.0934, 0.584]
when the drug was administered, indicating that the drug is effective in
ameliorating the disease phenotype. Since the Drug did not
completely eliminate the mutant phenotype, we need to calculate the net
effect of the drug.
Delta-delta comes in handy in this situation. We use the
Placebo delta as a reference for how much the mutant
phenotype is supposed to be, and we subtract the Drug delta
from it. The bootstrapped mean differences (delta-delta) between the
Placebo and Drug group are plotted at the
bottom right with a separate y-axis from other bootstrap plots. This
effect size, at about -0.903 [95% CI: -1.28, -0.513], represents the net
effect size of the drug treatment. In other words, treatment with drug A
reduced the disease phenotype by 0.903.
The mean difference between mutants and wild types given the placebo treatment is:
The mean difference between mutants and wild types given the drug treatment is:
The net effect of the drug on mutants is:
where is the sample mean, is the mean difference.
Specifying Grouping for Comparisons
In the example above, we used the convention of “test - control’ but
you can manipulate the orders of experiment groups as well as the
horizontal axis variable by setting experiment_label and
x1_level.
unpaired_delta2_specified.mean_diff <- load(df,
x = Genotype, y = Measurement,
experiment = Treatment, colour = Genotype,
delta2 = TRUE,
experiment_label = c("Drug", "Placebo"),
x1_level = c("M", "W")
) %>%
mean_diff()
dabest_plot(unpaired_delta2_specified.mean_diff)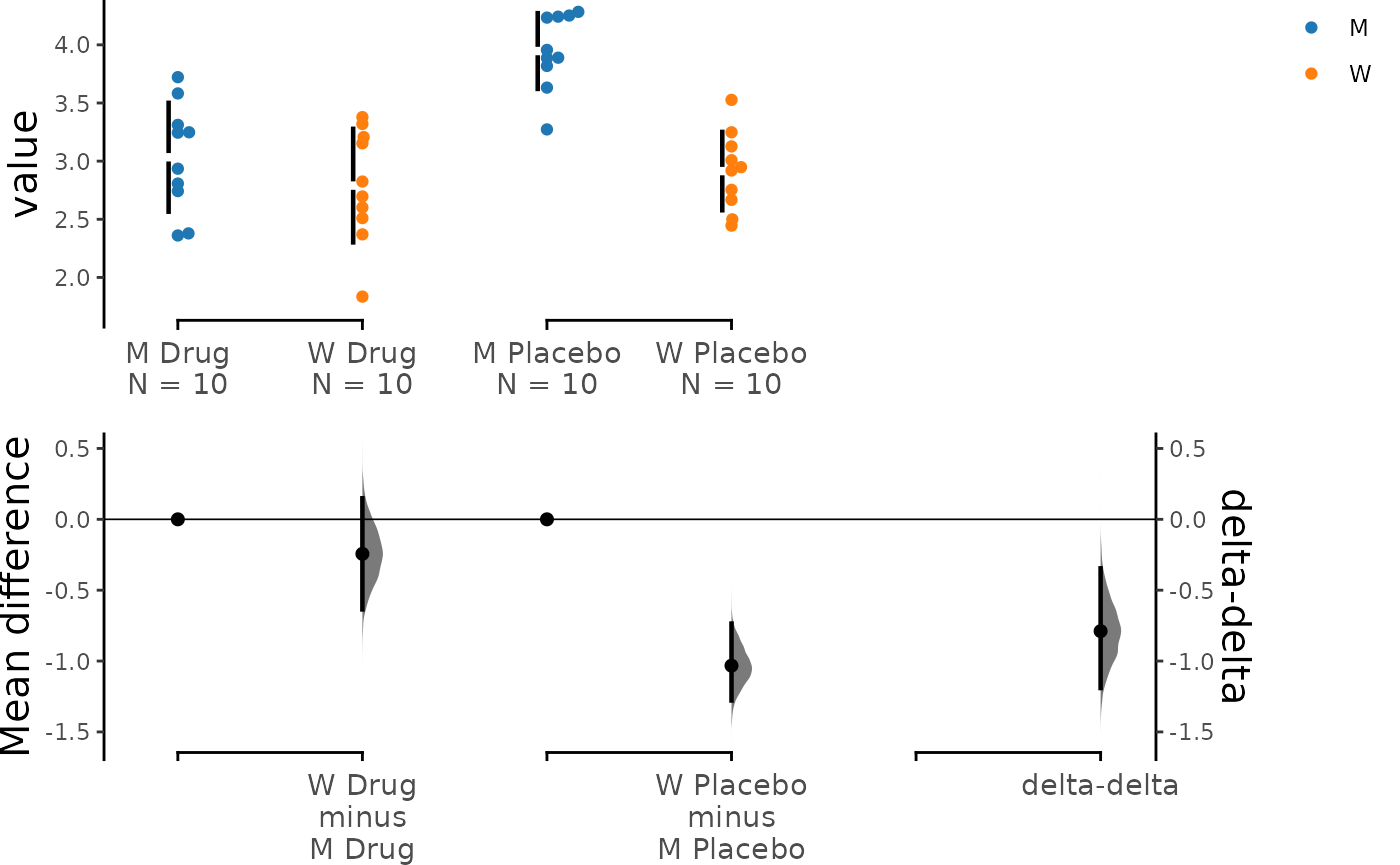
Paired Data
The delta-delta function also supports paired data,
which can be useful for visualizing the data in an alternative way. If
the placebo and drug treatment were administered to the same subjects,
our data is paired between the treatment conditions. We can specify this
by using Treatment as x and
Genotype as experiment. Additionally, we can
link data from the same subject with each other by specifying
ID as id_col.
Since we have conducted two replicates of the experiments, we can
colour the slope lines according to Rep to differentiate
between the replicates.
Although idx is an optional parameter, it can still be
included as an input to adjust the order of the data as opposed to using
experiment_label and x1_level.
paired_delta2.mean_diff <- load(df,
x = Treatment, y = Measurement,
experiment = Genotype, colour = Rep,
delta2 = TRUE,
idx = list(
c("Placebo W", "Drug W"),
c("Placebo M", "Drug M")
),
paired = "baseline", id_col = ID
) %>%
mean_diff()
dabest_plot(paired_delta2.mean_diff,
raw_marker_size = 0.5, raw_marker_alpha = 0.3
)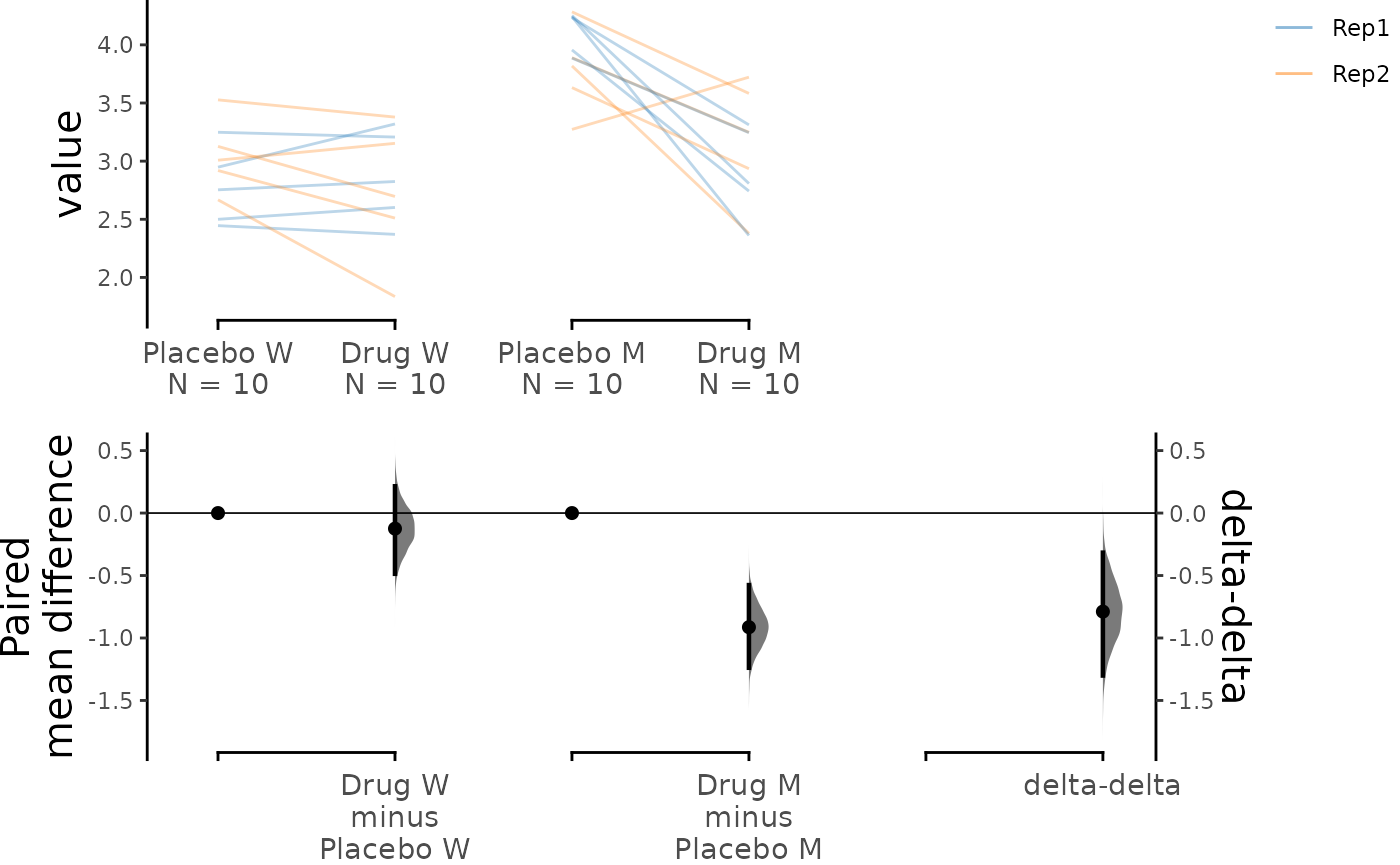
We see that the drug had a non-specific effect of -0.125 [95%CI -0.486 , 0.214] on the wild type subjects even when they were not sick, and it had a bigger effect of -0.913 [95%CI -1.24 , -0.577] in the mutant subjects. In this visualisation, we can see the delta-delta value of -0.789 [95%CI -1.3 , -0.317] as the net effect of the drug accounting for non-specific actions in healthy individuals.
The mean difference between drug and placebo treatments in wild type subjects is:
The mean difference between drug and placebo treatments in mutant subjects is:
The net effect of the drug on mutants is:
where is the sample mean, is the mean difference.
Connection to ANOVA
The comparison we conducted earlier is reminiscent of a two-way
ANOVA. In fact, the delta-delta is an effect size estimated for the
interaction term between Treatment and
Genotype. On the other hand, main effects of
Treatment and Genotype can be determined
through simpler, univariate contrast plots.
Omitting Delta-delta Plot
If for some reason you don’t want to display the delta-delta plot,
you can easily do so by setting show_delta2 to FALSE:
dabest_plot(unpaired_delta2.mean_diff, show_delta2 = FALSE)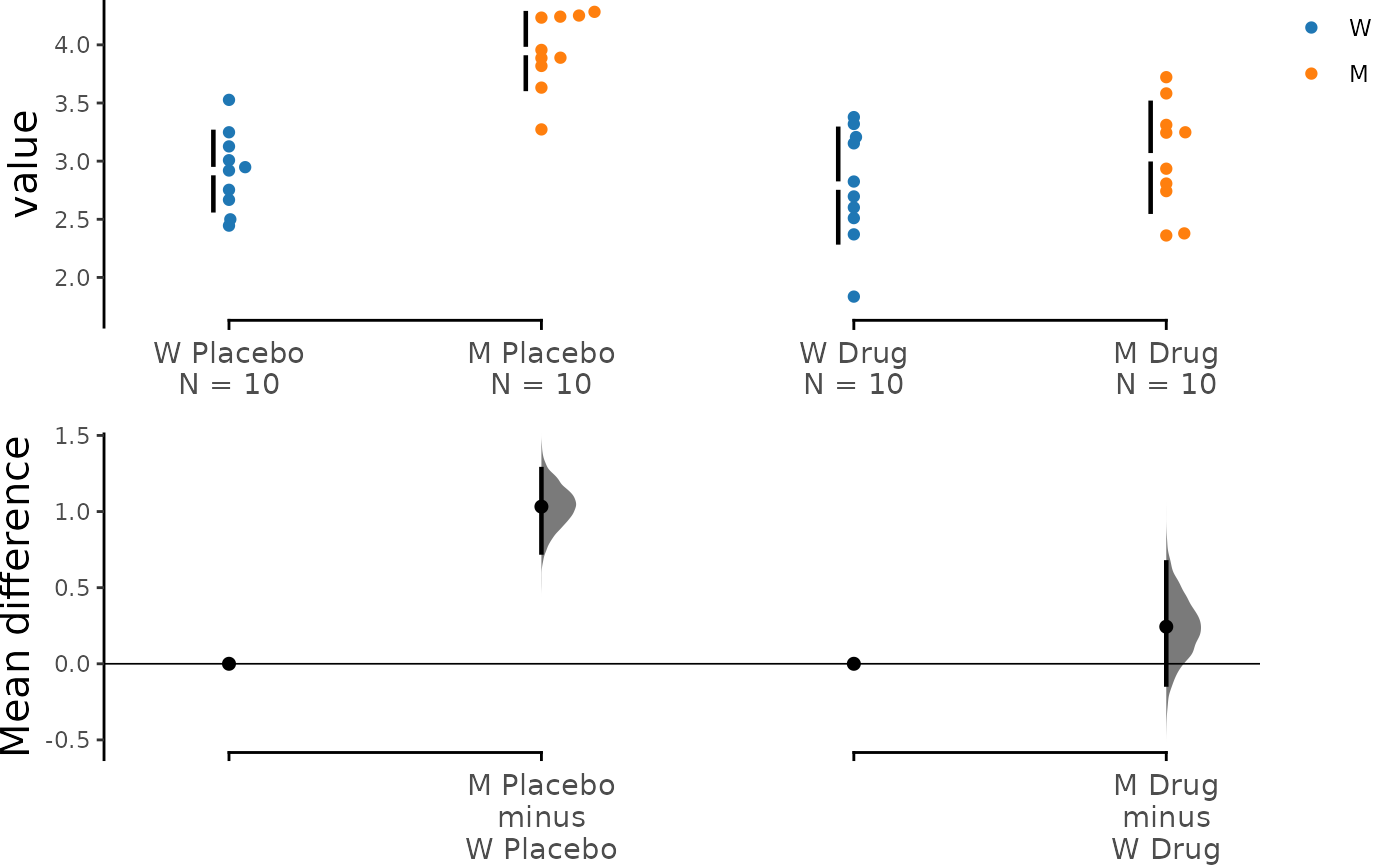
Other Effect Sizes
Since the delta-delta function is only applicable to mean differences, plots of other effect sizes will not include a delta-delta bootstrap plot.
# cohens_d(unpaired_delta2)Statistics
You can find all the outputs of the delta - delta calculation by
assessing the column named boot_result of the object
dabest_effectsize_obj.
print(unpaired_delta2.mean_diff$boot_result)
#> # A tibble: 3 × 11
#> control_group test_group bootstraps nboots bca_ci_low bca_ci_high pct_ci_low
#> <chr> <chr> <list> <int> <dbl> <dbl> <dbl>
#> 1 W Placebo M Placebo <dbl> 5000 0.731 1.28 0.748
#> 2 W Drug M Drug <dbl> 5000 -0.136 0.666 -0.151
#> 3 Delta2 Overall… Delta2 Ov… <dbl> 5000 -1.20 -0.337 -0.799
#> # ℹ 4 more variables: pct_ci_high <dbl>, ci <dbl>, difference <dbl>,
#> # weight <dbl>If you want to extract the permutations, permutation test’s p values,
the statistical tests and the p value results, you can access them using
the columns permutation_test_results,
pval_permtest, pval_for_tests and
pvalues respectively.
For instance, the P values for permutation tests
pval_permtest:
print(unpaired_delta2.mean_diff$permtest_pvals$pval_permtest)
#> [1] 0.0000 0.2628Or the permutation calculations and results could be accessed by:
print(unpaired_delta2.mean_diff$permtest_pvals$permutation_test_results)A representative p value for statistical tests
pval_for_tests:
print(unpaired_delta2.mean_diff$permtest_pvals$pval_for_tests)
#> $pvalue_welch
#> [1] 1.650881e-06
#>
#> $pvalue_welch
#> [1] 0.2694873Finally here the statistical test results and
pvalues:
print(unpaired_delta2.mean_diff$permtest_pvals$pvalues)
#> [[1]]
#> [[1]]$pvalue_welch
#> [1] 1.650881e-06
#>
#> [[1]]$statistic_welch
#> t
#> -6.971633
#>
#> [[1]]$students_t
#>
#> Welch Two Sample t-test
#>
#> data: control and test
#> t = -6.9716, df = 17.979, p-value = 1.651e-06
#> alternative hypothesis: true difference in means is not equal to 0
#> 95 percent confidence interval:
#> -1.3435832 -0.7212793
#> sample estimates:
#> mean of x mean of y
#> 2.914391 3.946822
#>
#>
#> [[1]]$pvalue_students_t
#> [1] 1.650881e-06
#>
#> [[1]]$statistic_students_t
#> t
#> -6.971633
#>
#> [[1]]$pvalue_mann_whitney
#> [1] 0.0002461281
#>
#> [[1]]$statistic_mann_whitney
#> W
#> 1
#>
#>
#> [[2]]
#> [[2]]$pvalue_welch
#> [1] 0.2694873
#>
#> [[2]]$statistic_welch
#> t
#> -1.139429
#>
#> [[2]]$students_t
#>
#> Welch Two Sample t-test
#>
#> data: control and test
#> t = -1.1394, df = 17.968, p-value = 0.2695
#> alternative hypothesis: true difference in means is not equal to 0
#> 95 percent confidence interval:
#> -0.6928931 0.2056392
#> sample estimates:
#> mean of x mean of y
#> 2.789728 3.033355
#>
#>
#> [[2]]$pvalue_students_t
#> [1] 0.2694873
#>
#> [[2]]$statistic_students_t
#> t
#> -1.139429
#>
#> [[2]]$pvalue_mann_whitney
#> [1] 0.3447042
#>
#> [[2]]$statistic_mann_whitney
#> W
#> 37