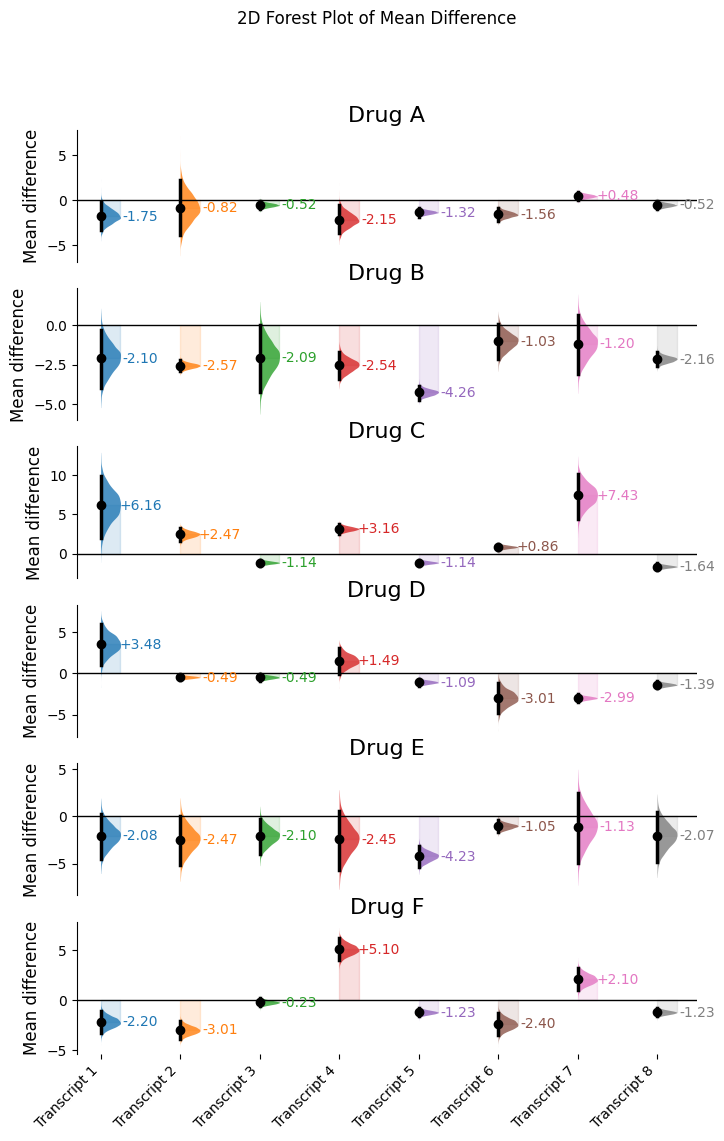
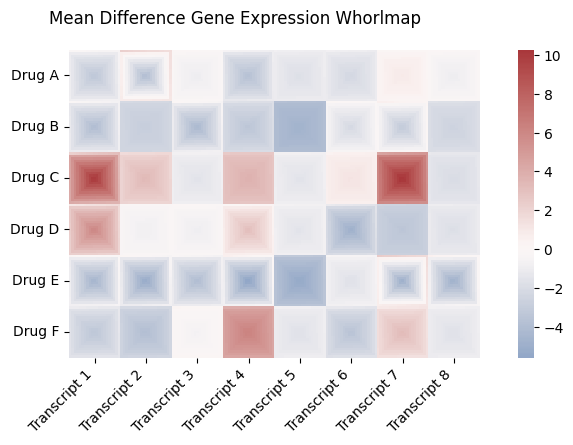
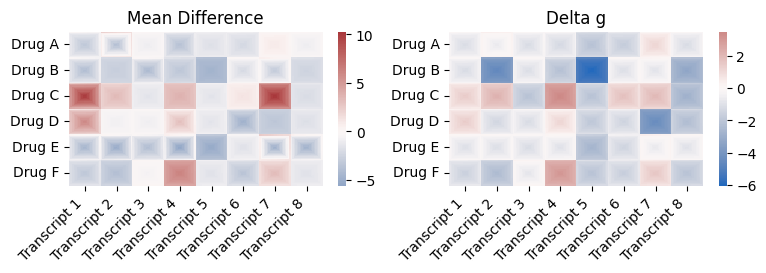
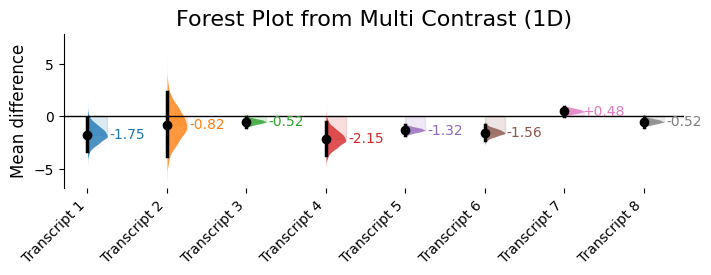
dabest_objects_2d = [[None for _ in range(8)] for _ in range(6)]
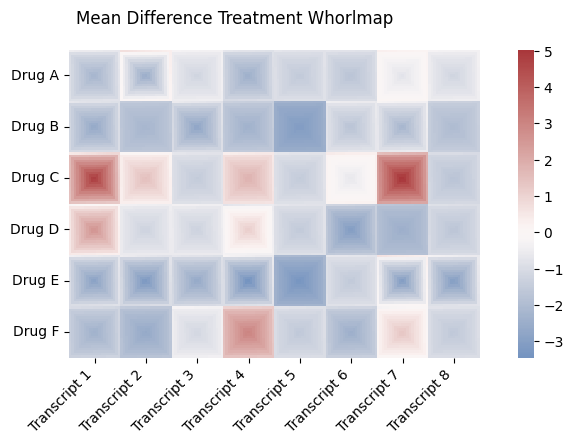
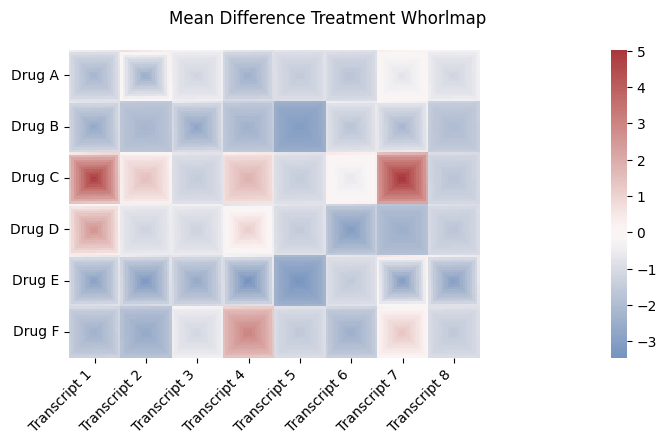
labels_2d = ["Transcript 1", "Transcript 2", "Transcript 3", "Transcript 4", "Transcript 5", "Transcript 6", "Transcript 7", "Transcript 8"]
row_labels_2d = ["Drug A", "Drug B", "Drug C", "Drug D", "Drug E", "Drug F"]
drug_effect_2d = [[.9, 2, 2, .5, 1.2, 1, 3,2, 3, 4],
[0.1, -.3, .1, -0.3, -2, 1.2, 1,.1,-4, 2],
[4, 4, 1, 5, 1, 3, 6.5,.5, -1.2, .4],
[6, 2, 2, 4, 1.4, -0.5, -.5,1.1, 3, .4],
[0.1, -.3, .1, -0.3, -2, 1.2, 1,.1,-4, 2],
[-.3, -1, 2, 7, 1, -0.5, 4,1, 2.3, -.4],
]
drug_effect_scale_2d = [[5, 10, 1, 5, 1, 2, 1,1, .1, 2],
[7, .2, 8, 3, 1, 4, 7,1, 5, 2],
[15, 3, 1, 2, 1, 1, 11,1, 7, 2],
[8, .1, 1, 5, 1, 6,1,1, 3, .4],
[9, 10, 7, 12, 4, 2,14,10, 9, 20],
[4, 3, 1, 4, 1, 4,4,1, 3, 4],
]
seeds = [1, 1000, 20, 9999, 1000, 5320]
for i in range(len(row_labels_2d)):
for j in range(len(labels_2d)):
df = create_delta_dataset(seed=seeds[i],
fourth_quarter_adjustment=drug_effect_2d[i][j],
scale4=drug_effect_scale_2d[i][j],
initial_loc = 20)
dabest_objects_2d[i][j] = dabest.load(data=df,
x=["Genotype", "Genotype"],
y="Transcript Level",
delta2=True,
experiment="Treatment")